








généralités
matières en suspension et colloïdes
définitions
On se reportera aux sections états des impuretés dans l'eau, les eaux souterraines et les eaux de mer et eaux saumâtres pour trouver des précisions et discussions sur les trois catégories d’impuretés dont l’élimination constitue l’objectif du traitement d’une eau :
- matières en suspension (sable, limons, plancton, débris organiques…) ;
- matières colloïdales (argiles fines, kystes de protozoaires, bactéries, macromolécules…) ;
- matières dissoutes (MO, sels, gaz…)
responsables, les deux premières de la turbidité, les deux dernières de la couleur, la dernière de la salinité et de diverses autres caractéristiques des eaux.
rôle de la coagulation-floculation
Les procédés de coagulation et de floculation facilitent l’élimination des MESet des colloïdes en les rassemblant sous forme de floc dont la séparation est ensuite effectuée par des systèmes de décantation, flottation et/ou filtration (voir Décantation et Filtration, de même que Floculateurs - décanteurs - flottateurs et Les filtres).
Ils constituent les traitements de base appliqués pour corriger tout ou partie des défauts de l’eau liés aux fractionsparticulaires inertes (limons, argiles, colloïdes) ou vivantes (microalgues planctoniques ; micro- invertébrés, en particulier les kystes des protozoaires parasites : amibes, Giardia, Cryptosporidium… ; bactéries) ; ils assurent aussi l’élimination de la fraction « floculable » des matières organiques (macromolécules, en particulier la plupart des acides humiques responsables de la couleur), de certains métaux lourds, plus généralement de la fraction des micropolluants associée à ces MES et macromolécules colloïdales (dont les virus, pratiquement toujours portés par les MES et colloïdes de l’eau).
les suspensions colloïdales – nécessité de la coagulation
stabilité des suspensions colloïdales
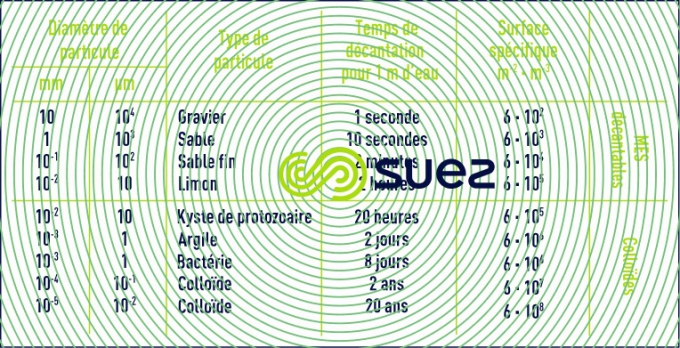
Dans le tableau 1 sont répertoriés certains matériaux ou organismes avec leur dimension et l’ordre de grandeur du temps nécessaire pour que, sous la seule influence de leur poids, ces particules parcourent verticalement un mètre d’eau à 20 °C.

(voir différents types de décantation)
Le tableau 1 montre donc que les colloïdes sont des particules :
- impossibles à décanter naturellement ;
- ayant une surfacespécifique très élevée qui régit la stabilité de leur suspension dans l’eau.
En effet, pour obtenir des vitesses de décantation plus rapides, il faudrait assembler un très grand nombre de colloïdes en agrégats d’au moins 10 à 100 mm, mais ces colloïdes exercent entre eux des forces de répulsion de nature électrostatique empêchant leur rapprochement : leur suspension peut donc rester parfaitement stable.
théorie de la double couche
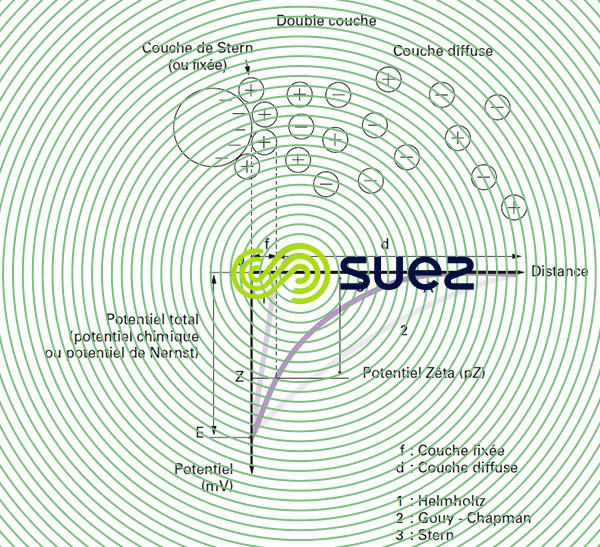
Les colloïdes présents dans l’eau brute sont très généralement chargés négativement (imperfections du réseau cristallin, ionisation des groupements chimiques périphériques…). Afin de neutraliser cette charge négative de surface, des ions positifs (appelés « contre-ions »), présents dans l’eau brute ou ajoutés, sont attirés et viennent former une couche autour du colloïde. Diverses théories ont été avancées (figure 1) :
- théorie de Helmholtz : Une couche d’ions positifs recouvre intégralement la surface du colloïde et assure la neutralité de l’ensemble (couche fixée) ;
- théorie de Gouy-Chapman : La couche d’ions positifs est inégalement répartie autour du colloïde ; la neutralité est obtenue à plus grande distance (couche diffuse) ;
- théorie de Stern qui combine les deux précédentes et considère la formation d’une double couche : la première formée d’ions du liquide mais adhérente au colloïde, la seconde diffuse dans le liquide environnant directement celui-ci. Comme illustré sur la figure 1 (courbe 3), le potentiel subit une première chute significative dans la couche fixée, puis diminue plus lentement à mesure que la distance augmente jusqu’à son annulation au point A (point isoélectrique).
le potentiel zêta
Un colloïde se caractérise donc par 2 potentiels (figure 1) :
- E : Potentiel thermodynamique, encore appelé potentiel de Nernst, présent à la surface même du colloïde mais non mesurable par des méthodes simples ;
- Z : Potentiel à la surface de la couche fixée, aussi appelé potentiel électrocinétique ou potentiel Zêta (pZ). Ce potentiel reste, comme déjà indiqué, négatif, les charges des ions de la couche fixée ne compensant pas les charges négatives de surface du colloïde. Il régit l’interaction mutuelle des colloïdes et peut être mesuré par électrophorèse ; en effet, quand un colloïde est soumis à un champ électrique, il atteint une vitesse telle qu’un équilibre s’établit entre la force électrique d’attraction vers l’anode et la force de frottement due à la viscosité du milieu. La relation liant cette vitesse (mobilité électrophorétique) et le potentiel Zêta est de la forme :



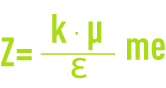
: Constante diélectrique du milieu,
μ : Viscosité dynamique (Pa · s),
k : fonction du diamètre de la particule et de l’épaisseur de la double couche.
On notera que, par définition, des particules ayant le même potentiel électrocinétique Zêta possèdent la même mobilité électrophorétique, indépendamment de leur diamètre.
L’appareil permettant la mesure du potentiel Zêta est le Zêtamètre (voir essais de traitabilité).
mécanisme de déstabilisation des suspensions colloïdales : la coagulation
Si deux particules colloïdales s’approchent l’une de l’autre, elles sont soumises à deux grands types de force de direction opposée (figure 2) :

c’est le second cas que l’on trouve dans les eaux naturelles, d’où la stabilité des suspensions colloïdales : on voit sur la partie droite de la figure 2 que l’évolution de la force résultante établit une « barrière énergétique » au voisinage des particules.

Pour déstabiliser la suspension (coagulation), il faut donc diminuer les forces de répulsion électrostatique, ce qui implique de neutraliser les charges superficielles des colloïdes : c’est ce qu’on obtient en ajoutant dans l’eau un produit dit « coagulant » (figure 3).
Dans la théorie de la double couche, la coagulation optimale peut être définie comme l’ajout de réactif permettant l’annulation du potentiel Zêta.

les étapes de l’agrégation
facteurs influençant la coagulation
La coagulation est donc la déstabilisation des particules colloïdales par addition d’un réactif chimique, le coagulant, qui apporte au milieu des cations multivalents, libres ou liés à une macromolécule organique (polyélectrolyte cationique). Ces cations sont adsorbés et fixés dans la première couche de Stern ; le pZ croît alors (figure 3) jusqu’à devenir nul ou négligeable lorsque la neutralisation de toutes les charges électronégatives de la particule est achevée (figure 7, essais de traitabilité).
On notera que pour être efficace, le coagulant doit être immédiatement dispersé dans l’eau pour obtenir une répartition homogène de celui-ci, et ceci avant toute précipitation d’hydroxyde. Il faut pour cela dissiper une énergie d’agitation importante pendant un temps court, ou en d’autres termes utiliser un gradient de vitesse très élevé.
En régime turbulent, le gradient de vitesse est défini par la formule :
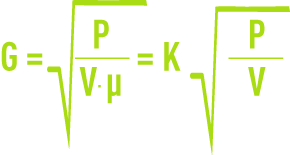
G : Gradient de vitesse moyen (s–1),
P : Puissance réellement dissipée (W),
V : Volume occupé par le fluide (m3),
µ : Viscosité dynamique (Pa · s).
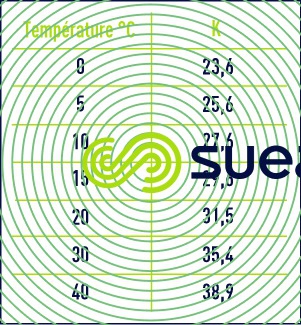
G dépend en particulier de la température via la constante K (tableau 2).
la floculation
C’est l’agglomération des particules (préalablement « déchargées ») en microflocs par pontage, soit par les hydroxydes résultant de l’hydrolyse du coagulant minéral, soit par les macromolécules du polyélectrolyte cationique. Les microflocs s’agrègent ensuite en flocons plus volumineux et décantables, le floc. Cette floculation peut être améliorée par l’ajout d’un autre réactif : l’adjuvant de floculation, plus simplement appelé le floculant.
En fait, ces agrégations successives composant le floc dépendent de deux phénomènes de transport qui régissent la vitesse de floculation :
- la floculation péricinétique liée à la diffusion brownienne (agitation thermique), où toutes les particules ont la même énergie cinétique et donc les plus petites ont les vitesses les plus élevées, d’où une plus grande probabilité de rencontre. La vitesse de floculation ou variation du nombre de particules agrégées au cours du temps est alors donnée par :
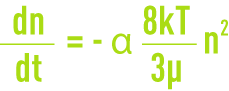
n : nombre de particules par unité de volume,
α : fraction des chocs efficaces,
k : constante de Boltzmann,
T : température absolue.
Cette loi n’est valable que pour de petites particules dont la taille est inférieure à 10 mm. Elle décrit la formation du microfloc, on y remarque l’influence de la « densité » de particules (n) et celle de la température ;
- la floculation orthocinétique liée à l’énergie mécanique dissipée dans la zone de floculation. L’efficacité de cette floculation qui permet d’obtenir un floc volumineux donc séparable est donnée dans le tableau 3.

On constate que le gradient de vitesse est aussi un paramètre très important de la vitesse de floculation.
En floculation, le gradient de vitesse agit sur la probabilité de rencontre des microflocs, mais il n’est pas possible de l’augmenter exagérément. En effet, pour des valeurs trop élevées de G, le floc formé subit un cisaillement mécanique entraînant sa destruction. Les valeurs généralement admises pour G sont :
- en coagulation : 400, voire 1 000 s–1 ;
- en floculation : de l’ordre de 100 s–1 et moins dès que le floc atteint une taille supérieure au millimètre.
temps de coagulation et de floculation
L’unité de temps de la coagulation est la seconde, tandis que celle de la floculation est la minute (ex. typique : 3 secondes - 20 minutes).
La mise en œuvre de la réaction de floculation peut être caractérisée par le paramètre adimensionnel G·ξ (ξ = temps de contact). La valeur de ξ peut être déterminée par un essai de floculation (voir mesure des paramètres globaux).
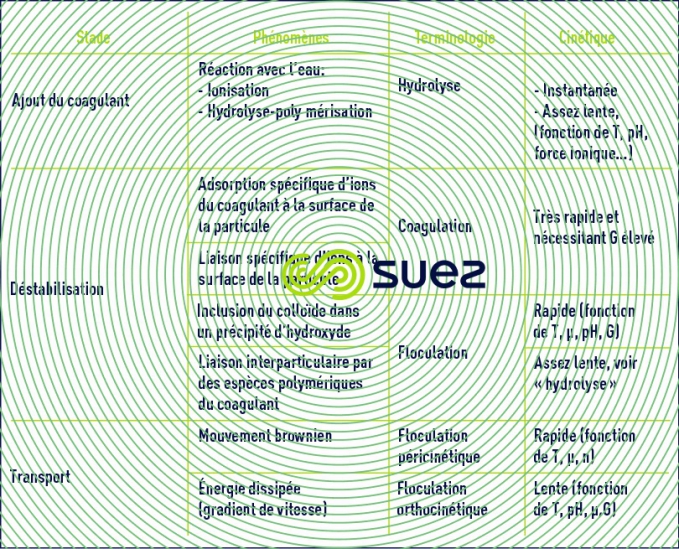
synthèse des phénomènes
Au total le tableau 4 récapitule les diverses phases successives ou simultanées conduisant à la formation de flocs à partir d’une suspension colloïdale.
les coagulants
cations trivalents
La coagulation est d’autant plus efficace que la valence du cation est élevée (théorie de Schulze-Hardy) :

z = valence du contre-ion utilisé ;
ainsi un ion trivalent est environ dix fois plus efficace qu’un ion divalent. Aussi, en pratique, les sels de fer ou d’aluminium trivalents ont été, et continuent d’être, largement utilisés dans tous les traitements de coagulation d’eau.
influence du pH
Les coagulants minéraux, par suite de leur hydrolyse, modifient les caractéristiques physico-chimiques de l’eau à traiter (pH, TAC, conductivité) :

Par ailleurs, le pH optimal constitue un compromis entre :
- le pH nécessaire à la coagulation (lié à la nature des colloïdes, leur point isoélectrique) ;
- le pH nécessaire à la floculation (lié à la croissance du floc d’hydroxyde de fer ou d’aluminium – tableau 5). Il correspond en général au minimum de solubilité de l’hydroxyde considéré (nécessaire aussi pour assurer le minimum de métal dissous dans l’eau, suivant les normes de potabilité en vigueur).

Ce pH et la solubilité minimale sont fortement influencés par la force ionique et la présence de composés organiques tels que les acides humiques.
Le pH de coagulation doit, au besoin, être ajusté par ajout d’acide ou de base.
taux de traitement
Le taux de traitement à mettre en œuvre est donné par un essai de floculation. Il peut être ajusté par l’étude du potentiel Zêta (voir intérêt des bioréacteurs à membranes).
La coagulation « idéale » correspond à pZ = 0, ce qui permet une élimination optimale de toutes les particules (turbidité argileuse, microalgues…) ; en traitement des eaux potables, l’élimination poussée des MO dissoutes peut demander un taux de traitement supérieur ; une dose de coagulant progressivement supérieure à celle qui annule le pZ entraîne d’abord la réapparition d’une turbidité résiduelle (figure 4, établie en unités arbitraires = UA) par inversion des charges (pZ rendu positif par les cations trivalents), puis à son élimination par piégeage des colloïdes dans le floc en excès (coagulation par entraînement) ; ce mode de coagulation est parfois appliqué conjointement avec un abaissement du pH (coagulation renforcée, appliquée en particulier pour une élimination poussée des précurseurs organiques de sous-produits d’oxydation).

À l’opposé, on utilise parfois le terme de « microfloculation » au sens de court temps de floculation, permettant de former un floc de petite taille, d’un diamètre de 1 à 2 mm, mais suffisant pour être séparé sur flottateur ou filtres. En filtration directe on peut utiliser conjointement un taux de coagulant réduit.
production de boue
La formation d’hydroxyde métallique entraîne la production d’un volume de boue important. Ces boues peuvent rarement être rejetées telles quelles dans le milieu naturel et doivent donc être traitées (voir Traitement des boues liquides et le traitement des boues).
coagulants organiques
Parfois utilisés à la place ou en complément des coagulants minéraux, ces polymères cationiques neutralisent directement par leurs charges positives les colloïdes négatifs. Leur adsorption à la surface de ceux-ci permet en même temps une action de pontage, donc de floculation. Un des avantages importants est que le volume des boues produites est considérablement réduit (absence d’hydroxyde).
les adjuvants de floculation (ou floculants)
Pour améliorer la floculation, des polymères minéraux (silice activée) ou naturels (amidon, alginate) ont d’abord été utilisés. Mais l’apparition de polymères de synthèse (très longues macromolécules ayant tendance à s’adsorber sur les microflocs et donc à les relier) a fait évoluer considérablement les performances de la floculation en permettant la formation de flocs plus gros et résistant mieux aux contraintes de cisaillement.
Le taux optimal de traitement à mettre en œuvre est donné, comme pour le coagulant, par l’essai de floculation (jar-test, voir dans analyses spécifiques) éventuellement complété par des essais de décantabilité des boues.
Le temps à respecter entre les ajouts du coagulant et du floculant est primordial : en effet, un floculant n’est efficace que lorsque la phase de microfloculation est achevée. Ce temps est fonction de divers facteurs (composition de l’eau, température…) et doit être déterminé expérimentalement dans chaque cas.
L’emploi de floculants de synthèse conduit à des volumes de boues inférieurs.
influence d’une préoxydation
Une préchloration (par exemple en eau de mer) et surtout une préozonation sont connues comme permettant de faciliter le mécanisme de coagulation-floculation. On peut invoquer ici la destruction ou du moins la désorption de la pellicule organique qui entoure certaines particules colloïdales et gêne ainsi la fixation des cations déstabilisants. En outre l’ozone à faible dose (< 1 g · m–3) permet une action de réduction du nombre de particules et d’augmentation de la masse molaire de certains composés organiques (réticulation), également favorable à la coagulation. C’est typiquement le cas des eaux riches en matières organiques et en algues, ou en matières organiques complexantes du fer ou du manganèse ; dans ce dernier cas, l’ozone détruit les complexes organiques et oxyde les ions métalliques ainsi libérés.



Outil Marque-page
Cliquez sur l'outil marque-page, puis surlignez le dernier paragraphe lu pour pouvoir poursuivre ultérieurement votre lecture.
